
With graph attention mechanism, a ShanghaiTech team created an explainable artificial intelligence (AI) molecular representation method that can help medicinal chemists gain chemical insights from relevant drug discovery data directly.
Hunting for chemicals with favorable pharmacological, toxicological, and pharmacokinetic properties remains a formidable challenge for drug discovery. To date, over 5000 molecular descriptors have been designed to characterize chemical meaning. The conventional machine learning approaches for QSAR/QSPR have revolved around feature engineering for these molecular descriptors, in which the goal is to select a subset of the relevant descriptors for use in model construction. Feature engineering is time-consuming and often not generalizable. Deep learning provides us with powerful tools to build predictive models that are appropriate for the rising amounts of data, but the gap between what these neural networks learn and what human beings comprehend is growing. Moreover, this gap (due to the “black box” nature of deep learning models) may induce distrust and restrict deep learning applications in practice.
A ShanghaiTech team developed an artificial intelligence for molecular representation that helps medicinal chemists gain chemical insights from data (Image credit: Zhaoping Xiong)
A joint team from ShanghaiTech and Shanghai Institute of Materia Medica of Chinese Academy of Sciences recently approached this challenge by developing an AI molecular representation that is interpretable, and may help medicinal chemists gain chemical insights from discovery data directly.
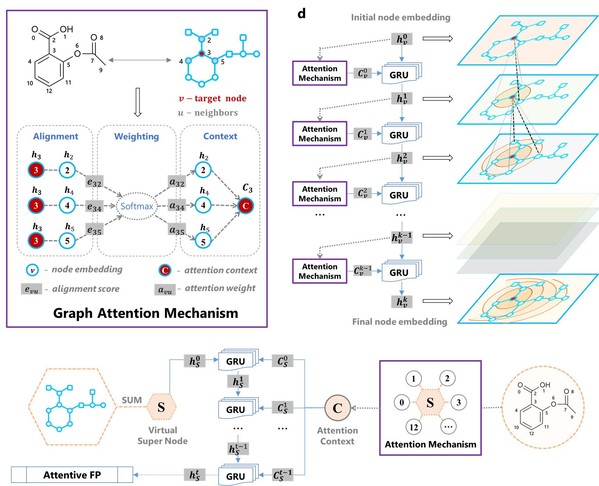
Called Attentive FP, this new molecular representation uses a graph attention mechanism to learn from relevant drug discovery related data. It achieves state-of-the-art predictive performances on a variety of data sets, and what it learns is interpretable via feature visualization for. The results suggested that Attentive FP automatically learns nonlocal intramolecular interactions from specified tasks, such as chemical environment and aromaticity.
“Efficient medicinal chemistry relies on associative reasoning and pattern recognition from molecular structures,” said Jiang Hualiang, professor of Shanghai Institute of Advanced Immunological Studies, “However, the use of empirical “drug-likeness” rules and “privileged” chemical (sub)structures is failing because the low-hanging fruit has become scarcer, which calls for more powerful molecular representation methods. Explainable AI is helpful to achieve accuracy as well as interpretability.”
Jiang says the research, selected as a cover story and online in the August 13 on Journal of Medicinal Chemistry, is an important step toward that ambitious goal by using explainable AI.
Other ShanghaiTech co-authors include graduate students Xiong Zhaoping and Liu Xiaohong. Funding for the research was provided by the National Natural Science Foundation of China.
This article originally appeared in Journal of Medicinal Chemistry.
Read more at:
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00959 (Paper link)
https://github.com/OpenDrugAI/AttentiveFP (AttentiveFP source code)