
近日,上海科技大学物质科学与技术学院黄焕明课题组通过自由基-极性交叉环加成反应(RPCC)实现了β-单取代以及β, β-二取代环状胺的多组分单步合成。该方法首次使用简单底物伯磺酰胺在同一反应中同时作为氮自由基前体和亲核试剂,发展了RPCC用于杂环化合物的合成新方法,为构建β取代环胺提供了新的逆合成思路。相关成果发表于国际化学领域代表期刊《德国应用化学》(Angewandte Chemie International Edition)。
环胺是合成化学中的基本组成部分。在美国FDA批准的1894种药物中,有59%的小分子药物含有环胺基序,并且哌啶和吡咯烷基序分别排名第1和第5。近年来,随着光催化氧化还原反应的突破,环状胺的自由基合成方法取得了长足发展,但通常仅局限于单组份或双组分反应,通过简单底物的多组分合成依旧非常困难。传统方法难以合成β取代的环状胺,且由于位阻效应,β, β-二取代的环状胺的合成往往更具有挑战性。

图 1 环状胺的常规逆合成思路
哌啶衍生物的常规合成方法包括吡啶的氢化还原、C-N键或C-C键形成、[4+2]环加成反应等(图1,左)。吡咯烷衍生物还可通过自由基环化、交叉偶联反应、[3+2]或[4+1]环加成反应得到(图1,右)。与之相比,RPCC方法可以从简单底物一步构建复杂结构,为杂环的高效构建提供了新的逆合成思路。
上海科技大学黄焕明课题组致力于发展可见光催化的自由基合成新方法研究。本研究开发了一种自由基-极性交叉环加成新方法,用于在温和的光氧化还原条件下从伯磺酰胺、未活化的烯烃和活化的烯烃合成环胺,反应有着出色的兼容性(图2,上)。此外,还可以使用伯磺酰胺、未活化的烯烃和催化量的芳基硫醇来生成β-单取代环胺(图2,下)。值得一提的是,该工作突破性地首次将伯磺酰胺同时作为氮中心自由基前体和亲核试剂。其提出的RPCC合成方法可以成为杂环合成的通用策略,也为伯磺酰胺作为双功能试剂带来了进一步发展潜力。
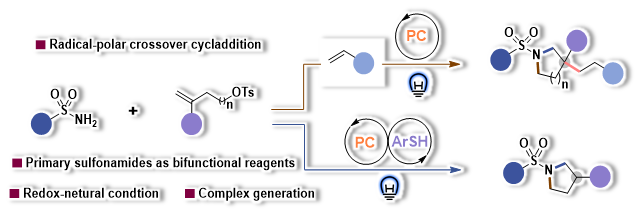
图 2 RPCC策略合成取代环胺